Болезнь Альцгеймера: роль генетических факторов: введение
Генетическая предрасположенность является четко установленным фактором риска болезни Альцгеймера. Более того, показано, что БА включает в себя несколько генетически гетерогенных форм, объединенных сходными клиническими и гистопатологическими признаками. Причиной или фактором риска развития некоторых (если не всех) форм БА являются мутации или полиморфизмы в ряде генов.
Прорывом в понимании этиологии БА явилось использование генетических подходов. Интенсивные эпидемиологические исследования около 10 лет назад позволили выявить родословные, в которых БА характеризуется ранним началом (моложе 65 лет) и моногенным аутосомнодоминантным характером наследования. При более распространенных поздних формах БА (у людей старше 65 лет) также очевидна роль генетических факторов, проявление которых, однако, зависит и от воздействий среды [ Pericak-Vance ea 1995 , Rao ea 1995 ]. К настоящему времени обнаружено, по крайней мере, четыре гена, мутации в которых вызывают БА или являются факторами генетической предрасположенности:
ген белка амилоидного предшественника (АРР) на хромосоме 21 [ Goate ea 1991 ],
ген аполипопротеина Е (АроЕ) на хромосоме 19 [ Sounders ea 1993 ] и
ген пресенилина-1 на хромосоме 14
ген пресенилина-2 на хромосоме 1 [ Sherrington ea 1995 , Rogaev ea 1995 , Levi-Lahad ea 1995 ].
Первый ген и генетический дефект, вызывающий болезнь Альцгеймера, был обнаружен с использованием подхода, который сейчас относят к так называемому "функциональному клонированию". Было давно отмечено, что синдром наиболее распространенной врожденной умственной отсталости, синдром Дауна , с трисомией по 21-й хромосоме, у пациентов старше 35-40 лет также характеризуется гистопатологическими признаками, характерными для БА. Из этого было сделано предположение, что ген (или гены) на 21-й хромосоме мог бы играть решающую роль в нейродегенеративном процессе также при БА.
Действительно, в конце 70-х годов был выделен один из основных компонентов амилоидных бляшек , которые накапливаются в неокортексе . Выделенный пептид, названный Ар , как оказалось, состоит всего из 39-43 аминокислотных остатков и является протеолитическим фрагментом более длинного пептида, названного предшественником амилоида ( АРР ) [ Kang ea 1987 , Lemaire ea 1989 ].
Ген АРР был локализован на хромосоме 21 , что сделало его явным кандидатом для поиска мутаций при болезни Альцгеймера. Действительно, вскоре были обнаружены несколько миссенс-мутаций в экзонах 16, 17, части которых кодируют Ар-пептид [ Goate ea 1991 , Murrel ea 1991 , Chartier-Harlin ea 1991 ].
Последующий массовый скрининг родословных и спорадических случаев во многих лабораториях мира показал, однако, что мутации в АРР могут объяснить причину БА только 3-5% всех родословных с ранним началом БА.
В родословных с поздним началом БА мутаций в этом гене не было найдено вовсе [ Tanzi ea 1992 , Schellenherg ea 1991 ].
Впоследствии было установлено, что полиморфизм (е4-аллель) еще в одном гене, аполипопротеине АроЕ , локализованном на хромосоме 19 , ассоциирован с некоторыми поздними и ранними формами БА [ Sounders ea 1993 , Poirier ea 1993 , Strittmater ea 1993 ]. Такой полиморфизм гена АроЕ, однако, не является необходимым или достаточным условием БА и рассматривается в настоящее время как фактор риска, влияющий на начало развития деменции альцгеймеровского типа [ Bennett ea 1995 ].
Предполагалось, что наиболее вероятными другими генами-кандидатами при БА могли бы стать гены, кодирующие пептиды протеолиза АРР белка, например, специфические а-, Р- или у-секретазы; белки, регулирующие синтез АРР; белки нейро-фибриллярных клубков (NFT) или киназы, регулирующие аномальное фосфорилирование отдельных компонентов NFT, например, tau- белков и т.д.
Значительным событием явилось недавнее обнаружение нового семейства генов, ответственных за БА, получивших название гены-пресенилины [ Sherrington ea 1995 , Rogaev ea 1995 ]. Важность открытия этих генов обусловлена тем, что были, во-первых, обнаружены принципиально новые, не предполагавшиеся ранее молекулярные компоненты нейропатологии БА, которые были выявлены "слепым" позиционным клонированием, не ориентированным на предыдущие "биохимические" модели болезни; во-вторых, идентифицированы генетические факторы, ответственные за наибольшее число случаев ранних семейных форм БА.
В начале 90-х годов для поиска новых хромосомных локусов, ответственных за болезнь Альцгеймера, был использован метод классического генетического сцепления с использованием случайных полиморфных маркеров ДНК. В значительной степени такой поиск был ускорен созданием генетических карт хромосом человека и накоплением нового поколения высокоинформативных микросателлитных маркеров (или STR -Simple Tandem Repeats).
Ряд таких маркеров, включая STR из хромосомы 14, были выделены к тому времени в лаборатории МГМ НЦПЗ РАМН в рамках проекта Геном Человека , как и в ряде других лабораторий [ Rogaev ea 1992 , Wang ea 1992 ].
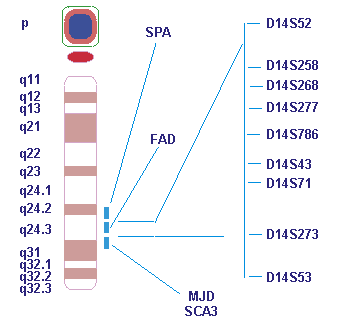
Результатом широкомасштабного поиска локуса БА явилось обнаружение в 1992 г. несколькими научными группами участка на хромосоме 14, ответственного за семейные формы БА ( FAD3 ). Мы использовали данные, полученные в Университете г. Торонто, по изучению более 20 семей с БА, в которых не было обнаружено сцепления БА с локусом АРР из 21-й хромосомы. После анализа более 70 маркеров из различных хромосом человека максимальный лод-балл и, следовательно, убедительное доказательство существования гена, мутации в котором ответственны за БА, были получены для маркера D14S43 (лод-балл > 20.47) из локуса 14q24.3 (локус FAD3) ( рис. 1 ). При этом было сделано два важных заключения. Во- первых, чрезвычайно высокий суммарный лод-балл, полученный при изучении совокупности семей различного этнического происхождения, свидетельствовал о том, что мутации в гене FAD3 ответственны за многие семейные формы БА (по крайней мере 60-70%) с ранним началом клинического проявления (до 60 лет). Во-вторых, результаты сегрегационного анализа STR-маркеров из локуса 14q24.3 показали, что FAD3 наследуется как моногенное аутосомнодоминантное заболевание, по всей видимости, с высокой пенетрантностью (проявлением мутаций), не зависящей от модификаций генетическими или эпигенетическими факторами.
В патогенезе по крайней мере некоторых форм болезни Альцгеймера участвуют и генетические факторы, например ген предшественника бета-амилоидного белка , расположенный на 21-й хромосоме . Так, у взрослых, страдающих синдромом Дауна ( трисомией по 21-й хромосоме ), после 40 лет развиваются характерные для болезни Альцгеймера патолого-анатомические нарушения. У многих из них формируется деменция , накладывающаяся на уже имеющуюся умственную отсталость . Полагают, что при синдроме Дауна накопление бета-амилоидного белка обусловлено активностью дополнительного гена предшественника бета-амилоидного белка . Кроме того, в единичных семьях с ранней формой семейной болезни Альцгеймера была выявлена точечная мутация в гене предшественника бета-амилоидного белка ; эти случаи послужили первым доказательством возможности моногенного аутосомно-доминантного наследования заболевания. Чаще всего мутация приводит к замене валина на изолейцин в положении 717.
Обследование семей, в которых болезнью Альцгеймера страдали несколько поколений, выявило два дополнительных гена, участвующих в патогенезе этого заболевания, - ген PSEN1 и ген PSEN2 .
Ген PSEN1 расположен на 14-й хромосоме и кодирует белок пресенилин-1 . Мутация этого гена вызывает болезнь Альцгеймера типа 3, которая наследуется аутосомно-доминантно с высокой пенетрантностью. В семьях с самой различной этнической принадлежностью выявлено более 20 мутаций гена PSEN1.
Ген PSEN2 расположен на 1-й хромосоме и кодирует белок пресенилин-2 . Мутация этого гена впервые была обнаружена у американских потомков немцев Поволжья.
Оба гена очень похожи и кодируют сходные белки. Сначала полагали, что эти белки имеют семь трансмембранных доменов, однако дальнейшие исследования показали, что число доменов равно восьми, а девятый располагается у внутренней стороны мембраны. Функция этих белков в норме, а также ее изменения, возникающие в результате мутации кодирующих их генов, неизвестны. Оба белка экспрессируются в нейронах и широко распространены в нервной системе . По первичной структуре они похожи на белок sel12 , участвующий в морфогенезе у нематоды Caenorhabditis elegans .
Мутации генов PSEN1 и PSEN2 сопровождаются повышением концентрации в плазме бета-амилоидного белка , состоящего из 42 аминокислотных остатков, что позволяет предположить взаимосвязь между пресенилинами и предшественником бета-амилоидного белка .
Доказано, что мутации гена PSEN1 - самая частая причина семейной болезни Альцгеймера с ранним началом, обусловливающая 70% случаев этой относительно редкой формы. Болезнь Альцгеймера типа 3, вызванная мутацией гена PSEN1 , характеризуется более ранним началом (в среднем в 45 лет) и более быстрым прогрессированием (средняя продолжительность жизни больного 6-7 лет), чем болезнь Альцгеймера типа 4, вызванная мутацией гена PSEN2 (начало в среднем в 53 года; средняя продолжительность жизни 11 лет). При некоторых редких мутациях гена PSEN2 заболевание начинается после 70 лет.
Роль мутаций этих генов в патогенезе гораздо более частых спорадических форм болезни Альцгеймера с поздним началом не изучена.
В настоящее время разработана методика исследования ДНК клеток крови, позволяющая выявлять такие мутации; пока она применяется только в исследовательских работах. Обследование необходимо при генетическом консультировании лиц с высоким риском заболевания, но без признаков деменции .
Важной вехой стало открытие роли гена, кодирующего апопротеин Е и расположенного на 19-й хромосоме , в развитии поздней семейной и спорадических форм болезни Альцгеймера. Апопротеин Е участвует в транспорте холестерина . Ген апопротеина Е имеет три аллеля: эпсилон2, эпсилонЗ и эпсилон4, кодирующие соответственно изоформы Е2, Е3 и Е4. Аллель эпсилон4 часто обнаруживают при спорадических и поздней семейной формах болезни Альцгеймера. Примерно 24-30% здоровых представителей белой расы имеют хотя бы один аллель эпсилон4, а около 2% - гомозиготны по нему. Среди больных болезнью Альцгеймера от 40 до 65% имеют по крайней мере один аллель эпсилон4 - иными словами, среди больных распространенность этого аллеля существенно выше, чем среди здоровых. С другой стороны, у многих больных аллель эпсилон4 отсутствует и, напротив, имеется у многих здоровых. То есть наличие аллеля эпсилон4 не является ни необходимым, ни достаточным условием болезни Альцгеймера, но служит важным фактором ее риска (особенно у гомозигот). Количество аллелей влияет на возраст начала заболевания: у гомозигот болезнь Альцгеймера начинается раньше. Причины этого не ясны.
Апопротеин Е присутствует в амилоидных бляшках ; он связывается с тау-белком и поэтому, возможно, участвует в образовании нейрофибриллярных включений . Апопротеин Е4 подавляет рост нервных окончаний в культуре клеток спинномозговых ганглиев.
Полагают, что аллель эпсилон2 уменьшает риск болезни Альцгеймера, но эти данные не подтверждены. По непроверенным сведениям, носители аллеля эпсилон4 менее чувствительны к лечению такрином . Есть также данные о том, что аллель A гена альфа1-антихимотрипсина повышает риск болезни Альцгеймера у носителей аллеля эпсилон4.
Единого мнения о целесообразности исследования апопротеина Е в диагностике болезни Альцгеймера нет. У здоровых по результатам этого исследования нельзя судить о риске болезни Альцгеймера, поскольку у многих носителей аллеля эпсилон4 она не развивается. Однако у некоторых гомозиготных носителей аллеля эпсилон4 при позитронно-эмиссионной томографии обнаруживают снижение скорости обмена веществ в коре головного мозга. Некоторые считают эти изменения предвестниками болезни Альцгеймера. Высказывалось предположение, что гомозиготное состояние по аллелю эпсилон4 у пожилого больного с деменцией с вероятностью 96-98% свидетельствует о болезни Альцгеймера. Однако лишь небольшая часть больных гомозиготны по аллелю эпсилон4, и даже у них необходимо исключить обратимые причины деменции. Тем не менее апопротеин Е остается единственным важным биохимическим маркером болезни Альцгеймера.
Продолжается изучение роли апопротеина Е в патогенезе заболевания и его диагностическое значение. Необходимо исследовать взаимосвязь апопротеина Е с другими деменциями.
Показано, что наличие аллеля эпсилон4 не связано с деменцией при болезни Паркинсона и болезни Крейтцфельдта-Якоба .
Смотрите также:
{kind=link}