Гликогеноз типа III: общие сведения
Гликогеноз типа III (недостаточность амило-1,6-глюкозидазы, лимитдекстриноз)
В основе гликогеноза типа III лежит недостаточность амило-1,6-глюкозидазы , отщепляющей от главной цепи гликогена его боковые цепи. Этот фермент вместе с фосфорилазой ответствен за полный распад гликогена ; при его недостаточности накапливается аномальный гликоген с укороченными боковыми цепями, напоминающий остаток декстрина .
Лимитдекстриноз наследуется как аутосомно-рецессивный признак и встречается у представителей разных этнических групп, но чаще среди евреев-сефардов (выходцев из Северной Африки). Ген амило-1,6-глюкозидазы расположен на хромосоме 1 ( 1р21 ). Найдено более 30 разных его мутаций, причем две мутации в 3-м экзоне ( 17delAG и Q6X ) типичны для гликогеноза IIlb (подтипа гликогеноза типа III ), при котором скелетная мускулатура остается интактной. Анализ ДНК и выявление специфических мутаций позволяют обнаруживать носительство дефекта и осуществлять нренатальную диагностику.
Недостаточность амило-1,6-глюкозидазы обусловливает гепатомегалию , гипогликемию , низкорослость , ту или иную степень миопатии и кардиомиопатию . Обычно поражается печень и поражаются мышцы и в этих случаях болезнь называют гликогенозом типа IIIa . Однако примерно у 15% больных в процесс вовлекается только печень, и тогда болезнь называют гликогенозом типа IIlb ( рис. 135.2 ).
Клинические проявления. В раннем детском возрасте болезнь практически неотличима от гликогеноза типа I ; в обоих случаях наблюдается гепатомегалия , гипогликемия , гиперлипопротеидемия и задержка роста . Иногда увеличивается и селезенка , но почки сохраняют нормальные размеры. Важно отметить, что у большинства больных с глиенозом типа III гепатомегалия и печеночные симптомы с возрастом становятся менее выраженными, а после полового созревания вообще исчезают. Тем не менее в отдельных случаях, особенно среди японцев, возможно развитие цирроза печени . Наблюдались и аденомы печени , причем среди больных французского происхождения их частота может достигать 25%. Злокачественная трансформация аденом нехарактерна, но у двух больных на последних стадиях цирроза печени развился печеночноклеточный рак . Поражение мышечной ткани (гликогеноз типа IIIа) в детском возрасте обычно незаметно, но к 30-40 годам становится очевидным, проявляясь медленно прогрессирующей слабостью мышц и повышенной утомляемостью . Электромиография (ЭМГ) обнаруживает распространенную миопатию ; нарушается и проведение возбуждения по нервам. Обычно находят гипертрофию желудочков сердца , но явное нарушение функции сердца встречается редко. В ряде случаев печеночные симптомы выражены столь незначительно, что болезнь диагностируется лишь в зрелом возрасте, когда возникают нервно-мышечные нарушения. Начальные проявления можно принять за болезнь Шарко-Мари-Тута . Нередко обнаруживается СПКЯ , хотя фертильность , как правило, не снижается.
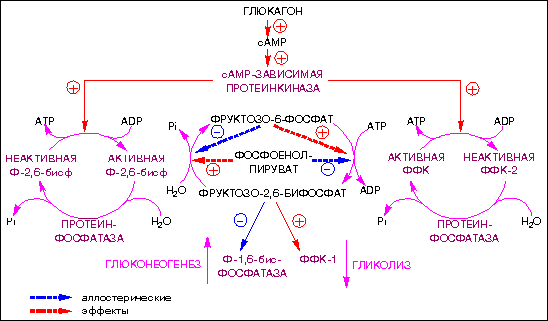
Обычно имеет место гипогликемия и гиперлипопротеидемия . В отличие от гликогеноза типа I , значительно возрастает активность печеночных трансаминаз и кетоновых тел ( кетоацидоз при голодании), но концентрация лактата и мочевой кислоты в крови остается нормальной. Введение глюкагона через 2 ч после завтрака с большим количеством углеводов вызывает нормальное повышение уровня глюкозы в крови, но после ночного голодания глюкагон уже почти не действует. О поражении скелетных мышц судят по активности КФК в сыворотке крови. Однако нормальный ее уровень не исключает недостаточности мышечной амилоло-1,6-глюкозидазы.
Диагностика. При гистологическом исследовании биоптатов печени видны переполненные гликогеном гепатоциты и фиброзные перегородки. Фиброз и малое содержание жира в печени отличают тип III гликогеноза от типа I. Фиброз может бьгть минимальным и захватывать лишь перипортальную область или достигать степени узелкового цирроза , но в большинстве случаев, по-видимому, не прогрессирует.
У больных с миопатией и печеночными симптомами (подтип IIIа) недостаточность амило-1,6-глюкозидазы обнаруживается не только в печени и мышцах, но и в других тканях - сердце, эритроцитах и в культуре фибробластов. В отсутствие клинических или лабораторных признаков миопатии недостаточность фермента ограничена печенью (подтип IIIb). Окончательный диагноз требует определения активности фермента в печени и/или мышцах. Неинвазивным диагностическим методом, позволяющим к тому же в большинстве случаев дифференцировать подтипы этого гликогеноза, является анализ мутаций.
Лечение. При гликогенозе типа III диетотерапия менее обязательна, чем при гликогенозе типа I. С гипогликемией, если она есть, обычно справляются путем частого кормления углеводной пищей с добавлением сырого кукурузного крахмала. Ночью крахмал вводят капельно через назогастральный зонд. Гипогликемию предотвращает также богатая белками диета (в дневные часы) и введение белковых растворов по ночам, поскольку аминокислоты служат субстратами глюконеогенеза, а этот процесс при гликогенозе типа III не нарушен. Надежных способов лечения прогрессирующей миопатии не существует. Больные не нуждаются в ограничении фруктозы и галактозы в диете. На конечных стадиях цирроза и/или при раке печени проводили трансплантацию этого органа.
Смотрите также: